
Tuesday, October 30, 2007
351 q3 moved to Friday
I won't make a habit of succumbing to the mob but I am moving the quiz to Friday. Hopefully this change doesn't screw anyone over. Tomorrow, Chapter 6, in which we will take thermo to a whole new level.
The Plight of Boltzmann+ Four Laws
 By the time the first lecture of Week 7 had finished, we had discussed all four laws of thermodynamics, in mathematical and verbal forms, established the probabilistic nature of entropy and worked through ten types of entropy calculations. In fact, nearly all of the framework has now been constructed for Chapter 6, in which everything comes to fruition as we discuss spontaneity and equilibrium.
By the time the first lecture of Week 7 had finished, we had discussed all four laws of thermodynamics, in mathematical and verbal forms, established the probabilistic nature of entropy and worked through ten types of entropy calculations. In fact, nearly all of the framework has now been constructed for Chapter 6, in which everything comes to fruition as we discuss spontaneity and equilibrium.Backtracking, last Friday we explored the work of [my idol] Ludwig Boltzmann, who beat his head against the scientific establishment as he spearheaded the development of statistical mechanics. Bypassing all the entropy is disorder nonsense, here we can see the physical underpinnings of entropy are probabilistic and that entropy is a measure of the number of accessible microstates to a system [which may arise as translational, rotational, vibrational, electronic, nuclear, configurational, etc]. In other words, entropy is a metric related to the number of ways that energy can be dispersed (into these microstates). Now that you have been equipped with this interpretation of absolute entropy, you can successfully point-and-laugh at all those who persist in utilizing the now-debunked disorder interpretation.
{kind=link}
{kind=link}
On Monday, we finished [finally] our set of ten processes:
01. cyclic process
02. reversible adiabatic
03. reversible isothermal
04. reversible phase change (at constant T, P)
05. reversible change of state [ideal gas]
06. irreversible change of state [ideal gas]
07. change of state [general] (two versions: T,V and T,P)
08. mixing of ideal gases A and B (also ideal solutions)
09. irreversible phase change (at constant T, P)
10. chemical reactions
These 10 processes cover nearly every situation of interest in chemistry.
Finally we elucidated the Third Law of Thermodynamics, that S → 0 as T → 0. As indicated in class, this is a restatement of what we saw in the Carnot engine, that absolute zero cannot be attained (although we've gotten way way down there, to 450 pK, where matter acts truly bizarre because of the dominance of quantum over thermal effects).
The absolute entropy of real matter, incidentally, usually approaches a nonzero S0, the residual entropy, which is a loose measurement of the strength of low-temperature intermolecular forces.
I hope it is clear by now that the Laws of Thermodynamics, in essence, establish a logical code from which nearly all energy transfer (and, hence, all phenomena) can be described. Turning this framework into usable results is not always easy, however.
Interestingly, a Fourth Law of Thermodynamics is often proposed, the Onsager reciprocal relations which we will not cover until pchem 2.
And now, the Four Laws of Thermodynamics, translated for Sanitation Engineers:
0th: There is shit.
1st: You can't get rid of it.
2nd: It gets deeper.
3rd: A nice empty trashcan is wishful thinking.
Thursday, October 25, 2007
The Second Law Is Better Than The Zeroth Law By Two Units
 On Wednesday, we finally made it to possibly the most powerful and darkly beautiful statement in all of thermodynamics: the Second Law. But, before that, we examined how we might calculate some entropy changes using the Clausius relation [dS = dqrev/T], underscoring how this equation only works while tracing reversible paths. The irreversible heat transfer from a hot to cold reservoir can, for example, be broken into three reversible -- and calculable -- steps. It is from this simple system that leads us to the 2nd Law conjecture:
On Wednesday, we finally made it to possibly the most powerful and darkly beautiful statement in all of thermodynamics: the Second Law. But, before that, we examined how we might calculate some entropy changes using the Clausius relation [dS = dqrev/T], underscoring how this equation only works while tracing reversible paths. The irreversible heat transfer from a hot to cold reservoir can, for example, be broken into three reversible -- and calculable -- steps. It is from this simple system that leads us to the 2nd Law conjecture:∆Suniv ≥ 0 [= for equil/reversible, > for spont/irreversible]Unfortunately, abuses of this statement are many and takes but a moment's googling to find them. For many scientists, it holds a special place:
The law that entropy always increases-the second law of thermodynamics-holds, I think, the supreme position among the laws of Nature. If someone points out to you that your pet theory of the universe is in disagreement with Maxwell's equations-then so much the worse for Maxwell's equations. If it is found to be contradicted by observation-well, these experimentalists do bungle things sometimes. But if your theory is found to be against the second law of thermodynamics I can give you no hope; there is nothing for it but to collapse in deepest humiliation." – Sir Arthur Eddington [1928]And to reiterate some general statements from class, which will hopefully help you more fully grasp spontaneity and reversibility:
All spontaneous processes are irreversible.Some common misstatements about the Second Law:
For all irreversible processes, ∆Suniv > 0.
All reversible processes are at equilibrium.
For all reversible processes, ∆Suniv = 0.
All systems tend to greater disorder.Before moving onto specific applications of the Clausius relation, we paused, for bookkeeping's sake, to mention the Zeroth Law: If systems A and B are in equilibrium, and systems B and C are in equilibrium, then systems A and C are in equilibrium. Not only does this establish that a state property common to them must be equal (the temperature), it also allows for the possibility that two systems could be in equilibrium without in direct contact.
False: Not only is "disorder" a poorly constructed idea and overly dependent on human interpretation, the underlying premise is wrong.
All systems tend towards greater entropy.
False: Some systems tend towards greater entropy while others don't. The restriction rests on ∆Suniv, not on ∆S.
Lastly we began our march of calculating entropy changes for ten processes, finishing four:
01. cyclic
02. reversible adiabatic
03. reversible isothermal
04. reversible phase transition [at const T, P]
On Friday, we will finish this list of ten, visit the revolutionary work of Boltzmann and, if time permits, elucidate the Third Law of Thermodynamics.
Tuesday, October 23, 2007
The Direction of Spontaneous Change
We began the first lecture of Week 6 with an examination of the inadequacy of the First Law to sufficiently describe thermodynamic events. For example, it does not preclude a penny, say, from absorbing thermal energy from a table and turning it into gravitational work (that is, springing up off the table). The Boltzmann formula will demonstrate that the probability of this event is nonzero but exceedingly small (so unimaginably improbable that perhaps we should call it impossible?) But the point is that, macroscopically, we see a definite directionality to energy transfer:

Through his theoretical work on heat engine efficiency, the French engineer Sadi Carnot was our first thermodynamicist. His memoirs, lost for twenty years and posthumously rescued by college friend Benoit Clapeyron, inspired the work of Clausius and Thomson [Kelvin], both of whom essentially triggered the thermodynamic revolution. Carnot's greatest achievement was to demonstrate that heat flow could be harnessed and transmogrified into usable work by engines, but with a maximum efficiency less than 100%. Indeed, this maximum efficiency is dependent only on the reservoir temperatures and not on the material used in the engine, nor on the actual steps of each cycle. It is a thermodynamic limit imposed on us by nature, who has decreed that heat is a form of energy rather than a transferred substance.
[Note: In today's lecture, I believe that I inadvertently flipped the subscripts for the temperatures in the adiabatic formulas. Consult Engel-Reid for consistency]
Looking further at the results of Carnot we see, as Clausius did, a hidden state function, one that sums to zero as we go around a cycle. From this fact we can back out the relationship dS = dq/T [the Clausius equation], introduced at the end of the hour but forming the basis of Wednesday's lecture to come.
First Law - limits the magnitude of energy transferBefore discussing how it was first discovered, we first needed to correct some misconceptions about entropy, one of the most thoroughly mangled concepts in all of science:
Second Law - limits the direction of energy transfer
Entropy is not equal to disorder, nor is it a measure of disorder (whatever that means scientifically).One of the most common [bad] examples demonstrating the alleged relationship between entropy and disorder is a deck of cards. When we shuffle an "ordered" deck of cards, we always see it become "disordered". The problem with this language is that there is no way to quantify order, especially since every outcome is equally probable. We have simply defined A,2,3,4 .. Q,K of each suit as being the ordered state -- but that definition is arbitrary. Nature should not -- and does not -- depend on such human definitions. Other [bad] examples include blaming messy desks and cluttered rooms on this "law of entropy."
When we connect it directly to the number of accessible microstates (ala Boltzmann) we will understand the probabilistic basis of entropy more fully.
Entropy is a measure of the tendency of energy to disperse, rather than being localized.

Through his theoretical work on heat engine efficiency, the French engineer Sadi Carnot was our first thermodynamicist. His memoirs, lost for twenty years and posthumously rescued by college friend Benoit Clapeyron, inspired the work of Clausius and Thomson [Kelvin], both of whom essentially triggered the thermodynamic revolution. Carnot's greatest achievement was to demonstrate that heat flow could be harnessed and transmogrified into usable work by engines, but with a maximum efficiency less than 100%. Indeed, this maximum efficiency is dependent only on the reservoir temperatures and not on the material used in the engine, nor on the actual steps of each cycle. It is a thermodynamic limit imposed on us by nature, who has decreed that heat is a form of energy rather than a transferred substance.
[Note: In today's lecture, I believe that I inadvertently flipped the subscripts for the temperatures in the adiabatic formulas. Consult Engel-Reid for consistency]
Looking further at the results of Carnot we see, as Clausius did, a hidden state function, one that sums to zero as we go around a cycle. From this fact we can back out the relationship dS = dq/T [the Clausius equation], introduced at the end of the hour but forming the basis of Wednesday's lecture to come.
Sunday, October 21, 2007
The Second Law
On Monday we will start Chapter 5 and move towards the Second Law of Thermodynamics, one of the most important statements in all of science. First I'll argue why the First Law is incomplete as a description of thermodynamics then dive headfirst into the Carnot cycle.
Wednesday, October 17, 2007
End of the First Law Era
How do we obtain reaction energies from reaction enthalpies, or vice versa? How do we determine a reaction enthalpy at a nonstandard temperature given a value at 298K? These two questions take us to the end of Chapter Four and the "First Law Era".
It is straightforward to derive the relation ∆rxnU°=∆rxnH°-RT∆rxnνgas which can be used to interconvert between reaction energy and enthalpy. When using this equation, we must keep in mind that (a) we are implicitly assuming all gases are ideal and (b) that, for liquids and solids, molar enthalpies and internal energies are approximately equal. In principle, we could jam in a real gas equation of state and create a ghastly version that is more general or, as is always done, use this version anyway and take the hit in accuracy. Assumption (b) is rather good because, unless we encounter extreme pressures, the heat capacities CP,m and CV,m are nearly equal for condensed phases.
Adjusting to a nonstandard temperature is rather important since many (most?) reactions do not actually occur at 25°C and the difference is often quite significant. Once the heat capacity CP,m is known as a function of temperature, we can calculate ∆∆rxnH°=∫∆rxnC°PdT.
To make integration life easier, the heat capacities are fit to simple polynomials of T:
CP,m=a + bT +cT2+dT3+eT4 [Shomate] or
CP,m=a + bT +cT-2
Exam 1 on Thursday. If you are nervous, just remember that if you got through organic chemistry, calculus and physics, you can do this. I have posted solutions to the two questions I assigned from Chapter 4. I also updated the study sheet that had some equations missing and an error in one of the thermodynamic equations of state.
It is straightforward to derive the relation ∆rxnU°=∆rxnH°-RT∆rxnνgas which can be used to interconvert between reaction energy and enthalpy. When using this equation, we must keep in mind that (a) we are implicitly assuming all gases are ideal and (b) that, for liquids and solids, molar enthalpies and internal energies are approximately equal. In principle, we could jam in a real gas equation of state and create a ghastly version that is more general or, as is always done, use this version anyway and take the hit in accuracy. Assumption (b) is rather good because, unless we encounter extreme pressures, the heat capacities CP,m and CV,m are nearly equal for condensed phases.
{kind=link}
Adjusting to a nonstandard temperature is rather important since many (most?) reactions do not actually occur at 25°C and the difference is often quite significant. Once the heat capacity CP,m is known as a function of temperature, we can calculate ∆∆rxnH°=∫∆rxnC°PdT.
To make integration life easier, the heat capacities are fit to simple polynomials of T:
CP,m=a + bT +cT2+dT3+eT4 [Shomate] or
CP,m=a + bT +cT-2
Exam 1 on Thursday. If you are nervous, just remember that if you got through organic chemistry, calculus and physics, you can do this. I have posted solutions to the two questions I assigned from Chapter 4. I also updated the study sheet that had some equations missing and an error in one of the thermodynamic equations of state.
Tuesday, October 16, 2007
Thermochemistry and Hess' Law
The first lecture of Week 5 found us tackling thermochemistry, that subset of thermodynamics describing heat transfer that accompanies chemical reactions. In constant-volume calorimeters (often closed vessels), the heat transfer q we measure is ∆rxnU, whereas in constant-pressure calorimeters (open vessels), q will be ∆rxnH. (On Wednesday, we will see a simple method to interconvert them). Since reactions are typically performed at constant temperature and pressure, any results give us important information about the energy stored in chemical bonds.
Using the properties of state functions, we can predict the heat transfer under these two conditions using Hess' Law and, since constant-pressure conditions are more common in chemical systems, we tend to focus on ∆rxnH rather than ∆rxnU. The norm is to cast all reactions as simple sums of formation reactions, each of which represents the formation of 1 mole of a substance from constituent elements in their standard states/phases. Hess' Law is particularly powerful in thermochemistry because it applies equally well for any extensive state property. Note that this is our second usage of the ∆ symbol (the first being the familiar ∆Y = Yfinal - Yinitial). Whenever the subscript appears on the ∆ itself, as in ∆combY, we are calculating the sum of the products minus the sum of the reactants, each multiplied by appropriate stoichiometric coefficients. Hopefully this operation is still familiar from general chemistry.
Next lecture, we will finish thermochemistry and begin to tackle entropy, one of the most important and poorly understood concepts in all of science. If your intrepid instructor has the backbone to trudge through the quiz 2 carnage, he may be able to return them on Wednesday. I will admit that the uncharacteristic dearth of questions/comments/emails/office visits so far this quarter (quickly approaching the 50%-done mark) had lulled me to mistakenly believe that this class was further along the thermodynamic path than it actually was. Hopefully quiz 2 will be a valuable learning experience for many as we lumber towards Thursday (and remember, no class on Friday).
Using the properties of state functions, we can predict the heat transfer under these two conditions using Hess' Law and, since constant-pressure conditions are more common in chemical systems, we tend to focus on ∆rxnH rather than ∆rxnU. The norm is to cast all reactions as simple sums of formation reactions, each of which represents the formation of 1 mole of a substance from constituent elements in their standard states/phases. Hess' Law is particularly powerful in thermochemistry because it applies equally well for any extensive state property. Note that this is our second usage of the ∆ symbol (the first being the familiar ∆Y = Yfinal - Yinitial). Whenever the subscript appears on the ∆ itself, as in ∆combY, we are calculating the sum of the products minus the sum of the reactants, each multiplied by appropriate stoichiometric coefficients. Hopefully this operation is still familiar from general chemistry.
Next lecture, we will finish thermochemistry and begin to tackle entropy, one of the most important and poorly understood concepts in all of science. If your intrepid instructor has the backbone to trudge through the quiz 2 carnage, he may be able to return them on Wednesday. I will admit that the uncharacteristic dearth of questions/comments/emails/office visits so far this quarter (quickly approaching the 50%-done mark) had lulled me to mistakenly believe that this class was further along the thermodynamic path than it actually was. Hopefully quiz 2 will be a valuable learning experience for many as we lumber towards Thursday (and remember, no class on Friday).
Monday, October 15, 2007
exam 1 on the horizon
The pchem fun will commence Thursday evening, 6:30 pm in Fisher Science North [53-213]. I will try to create something worthy enough for you and your finely honed instruments of pchem carnage...
Friday, October 12, 2007
Heat Capacity Difference & the Joule-Thomson Effect
Previously in this course we pulled from the thermodynamic heavens that, for an ideal gas, CP - CV = nR. Our first task in today's lecture was to find an expression for the heat capacity difference of any system and, in doing so, we showed from where the ideal gas relationship arises. Midstream in the derivation we paused to explain why CP and CV values are nearly identical for condensed phases (solids and liquids) under normal conditions, another result that was simply asserted before. Remember that the goal of pchem is to explain all of chemistry using simple models, so we like to pull relations from nowhere as rarely as possible.
In Wednesday's lecture, we had expanded on how the internal energy U varies with T and V, obtaining the internal pressure concept [(∂U/∂V)T] in the process. Today we performed an analogous treatment of the enthalpy H, finding how it varies with T and P, making it the third time this quarter we've associated U with V and H with P. Engel-Reid does not use this terminology but our results (Equations 3.20, 3.44) are commonly called the thermodynamic equations of state. Note that these are different from plain-vanilla equations of state, which tie together physical properties of a system, like P, T and V. The derivative (∂H/∂P)T will be rather important when we consider reactions and other processes that do not occur at 1 bar (for example, reactions occuring in the troposphere or several miles into the earth's mantle).
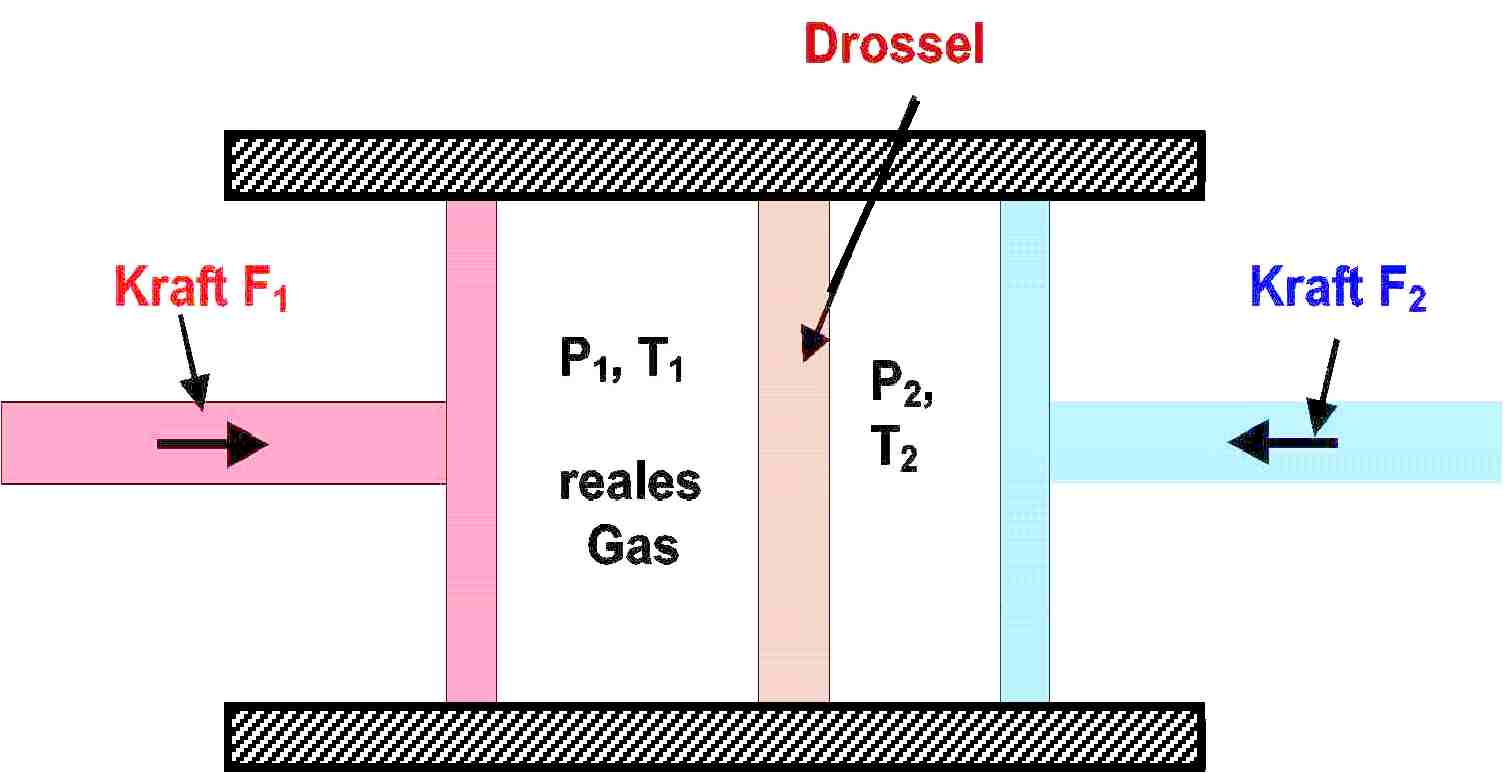
As we finish up Chapter 3, we then discussed the Joule-Thomson effect and coefficient, μT=(∂T/∂P)H, which measure the temperature response of a substance (usually a gas) to changes in pressure at constant enthalpy. Our intuition suggests that gases cool upon expansion, which is usually true and can be seen explicitly from the ideal gas law. But real gases are often unpredictable and several (like hydrogen and helium) have negative JT coefficients at standard conditions, meaning that they increase in temperature upon expansion, which is particularly important when handling tanks of hydrogen gas. The throttling apparatus devised by Joule and Thomson to attain isenthalpic conditions (in which no heat is extracted from nor no net work is done on the system) is particularly clever.
Monday will bring us to the [hopefully] familiar topic of thermochemistry [Chapter 4, which we will finish on Wednesday] and quiz 2, with exam 1 looming just over the horizon.
In Wednesday's lecture, we had expanded on how the internal energy U varies with T and V, obtaining the internal pressure concept [(∂U/∂V)T] in the process. Today we performed an analogous treatment of the enthalpy H, finding how it varies with T and P, making it the third time this quarter we've associated U with V and H with P. Engel-Reid does not use this terminology but our results (Equations 3.20, 3.44) are commonly called the thermodynamic equations of state. Note that these are different from plain-vanilla equations of state, which tie together physical properties of a system, like P, T and V. The derivative (∂H/∂P)T will be rather important when we consider reactions and other processes that do not occur at 1 bar (for example, reactions occuring in the troposphere or several miles into the earth's mantle).
As we finish up Chapter 3, we then discussed the Joule-Thomson effect and coefficient, μT=(∂T/∂P)H, which measure the temperature response of a substance (usually a gas) to changes in pressure at constant enthalpy. Our intuition suggests that gases cool upon expansion, which is usually true and can be seen explicitly from the ideal gas law. But real gases are often unpredictable and several (like hydrogen and helium) have negative JT coefficients at standard conditions, meaning that they increase in temperature upon expansion, which is particularly important when handling tanks of hydrogen gas. The throttling apparatus devised by Joule and Thomson to attain isenthalpic conditions (in which no heat is extracted from nor no net work is done on the system) is particularly clever.
{kind=link}
Monday will bring us to the [hopefully] familiar topic of thermochemistry [Chapter 4, which we will finish on Wednesday] and quiz 2, with exam 1 looming just over the horizon.
Thursday, October 11, 2007
P07 on hw.3
There is a typo in P07 on hw.3, which is Engel-Reid problem P3.8. The question should be asking for V as a function of T [not P] and beta, which should likewise be assumed to be independent of temperature.
Wednesday, October 10, 2007
Mathemagics
In today's lecture we introduced our last mathematical identity for partial derivatives, the cycle rule (also called the triple product rule and the cyclic chain rule). It will be employed in several contexts this quarter, but one useful application is expressing a single partial derivative in terms of the product of two others. Surprising relationships often arise when using this rule (which is valid for any three arbitrary thermodynamic state functions). In some mathematical derivations, it is especially helpful for getting rid of a variable that is difficult to hold constant, such as H or U.
Several thermodynamic partial derivatives go by special names (we have already met the heat capacities CP and CV). Today we discussed the thermal expansion coefficient, the isothermal compressibility and the internal pressure (all, in principle, functions rather than values).
Given an equation of state, we could analytically find functions for these parameters simply by calculating the partial derivative. On the other hand, since every thermodynamic partial derivative implies an experiment, we could also uncover these relationships in the lab when an equation of state is not available (which is most systems except gases). Tables 3.1 and 3.2 show some experimental results at standard conditions for various solids and liquids. (Note that Table 3.1 is mislabeled as isothermal coefficient rather than thermal expansion coefficient).
To further clarify what the internal pressure parameter is actually measuring, we calculated it for both an ideal gas and a van der Waals gas. As expected, since the internal energy U of an ideal gas is a function of T only, its internal pressure is zero. Another way of looking at it: For a gas' internal energy to be altered by changing the volume, that gas would necessarily possess intermolecular forces between its particles. The vdW gas gave a nonzero answer, the correction term for the pressure in the van der Waals equation!
Now that the Box of Mathemagics has been assembled, onward to more relations! Friday will see us relating CP and CV for any arbitrary substance and hopefully mine won't be the only tears of mathematical joy being shed...

Several thermodynamic partial derivatives go by special names (we have already met the heat capacities CP and CV). Today we discussed the thermal expansion coefficient, the isothermal compressibility and the internal pressure (all, in principle, functions rather than values).
Given an equation of state, we could analytically find functions for these parameters simply by calculating the partial derivative. On the other hand, since every thermodynamic partial derivative implies an experiment, we could also uncover these relationships in the lab when an equation of state is not available (which is most systems except gases). Tables 3.1 and 3.2 show some experimental results at standard conditions for various solids and liquids. (Note that Table 3.1 is mislabeled as isothermal coefficient rather than thermal expansion coefficient).
To further clarify what the internal pressure parameter is actually measuring, we calculated it for both an ideal gas and a van der Waals gas. As expected, since the internal energy U of an ideal gas is a function of T only, its internal pressure is zero. Another way of looking at it: For a gas' internal energy to be altered by changing the volume, that gas would necessarily possess intermolecular forces between its particles. The vdW gas gave a nonzero answer, the correction term for the pressure in the van der Waals equation!
Now that the Box of Mathemagics has been assembled, onward to more relations! Friday will see us relating CP and CV for any arbitrary substance and hopefully mine won't be the only tears of mathematical joy being shed...
Monday, October 8, 2007
State Functions, Euler's Criterion
After welcoming in Week 4 today, we finished our cyclic process question: isothermal expansion followed by adiabatic compression, ending in isochoric cooling. Once again we saw, through calculation rather than assertion, that q and w are path functions -- and nonzero -- while U and H are state functions, making ∆U=∆H=0 for the cycle. In the process we found that this area was negative, giving positive work (done on system by surroundings).
Our second problem addressed how we might calculate ∆H for any case in which the heat capacity is temperature-dependent. As in the ideal gas case, we can simply integrate the expression dH=CPdT; but unlike that example, the heat capacity is not constant. Still, the procedure is nearly identical. This is an important step towards generalizing our thermodynamic approach to systems beyond ideal gases.
Chapter 3 lays much of the mathematical foundation for the rest of the quarter. Hopefully you are beginning to see the importance of state functions and we will build heavily on this idea. But before today, I had simply asserted which properties were state functions and which were path functions. Now, with the introduction of Euler's Criterion for exactness, we have a mathematical litmus test: Iff the differential dZ is exact, then Z is a state function. Later, we will turn this principle on its head (in the form of Maxwell's relations) and generate a handful of remarkable thermodynamic relations that are far from obvious.
On Wednesday, backwards sixes will fly as we begin to build our Big Box of Mathemagics.
Our second problem addressed how we might calculate ∆H for any case in which the heat capacity is temperature-dependent. As in the ideal gas case, we can simply integrate the expression dH=CPdT; but unlike that example, the heat capacity is not constant. Still, the procedure is nearly identical. This is an important step towards generalizing our thermodynamic approach to systems beyond ideal gases.

Chapter 3 lays much of the mathematical foundation for the rest of the quarter. Hopefully you are beginning to see the importance of state functions and we will build heavily on this idea. But before today, I had simply asserted which properties were state functions and which were path functions. Now, with the introduction of Euler's Criterion for exactness, we have a mathematical litmus test: Iff the differential dZ is exact, then Z is a state function. Later, we will turn this principle on its head (in the form of Maxwell's relations) and generate a handful of remarkable thermodynamic relations that are far from obvious.
On Wednesday, backwards sixes will fly as we begin to build our Big Box of Mathemagics.
Sunday, October 7, 2007
This Forum
I decided to start this blog because it seemed like a good, central spot for course information, questions and clarifications about lecture/homework and a place to expand on or summarize things we do in class. I've never used this type of forum for a class before so I have no idea if it is even useful or worth my effort. So far, no one has asked any questions or added any comments -- maybe it's because we aren't far enough long or maybe this isn't the right venue. Maybe it is just easier to watch than participate. Maybe it's because reading/participation isn't assigned or I haven't placed any point values on it (which is against my philosophy, incidentally).
I've decided to keep this blog going until at least Exam 1 and then will reevaluate whether it is worthwhile. An interesting sidenote is that I've noticed from my webcounter that lots of students from other universities seem to be accessing information from this site. Perhaps that is meritorious in its own right.
I've decided to keep this blog going until at least Exam 1 and then will reevaluate whether it is worthwhile. An interesting sidenote is that I've noticed from my webcounter that lots of students from other universities seem to be accessing information from this site. Perhaps that is meritorious in its own right.
Saturday, October 6, 2007
Isobars, Isochores, Isotherms and Adiabats
 In Friday's lecture, we finished our uberproblem, obtaining expressions for q, w, ∆U and ∆H for the following reversible ideal gas processes: isobaric, isochoric, isothermal and adiabatic. Not only are the equations themselves useful, they demonstrate something that had previously been simply asserted, that q and w are path-dependent while ∆U and ∆H are not. We also saw (for the second time this quarter) the connections between U and constant-volume and H and constant-pressure conditions.
In Friday's lecture, we finished our uberproblem, obtaining expressions for q, w, ∆U and ∆H for the following reversible ideal gas processes: isobaric, isochoric, isothermal and adiabatic. Not only are the equations themselves useful, they demonstrate something that had previously been simply asserted, that q and w are path-dependent while ∆U and ∆H are not. We also saw (for the second time this quarter) the connections between U and constant-volume and H and constant-pressure conditions.It is important, however, to realize that these results are secondary to the calculation process itself. That is, what we get as the answer is somewhat less important than how we get it. Though we are focusing on the ideal gas as our system, the logic underlying these calculations apply just as readily to real gases and other substances, though the actual mathematics will be a bit messier.
We also developed some mathematical relations important to adiabatic processes and saw how the heat capacity ratio naturally arises in those equations, which tie together two of the three simultaneously changing variables (P, V, T).
On Monday, a new homework set [hw.3] as we venture into Chapter 3 and its elegant elegance.
Wednesday, October 3, 2007
Equipartition, Internal Energy, Heat Capacity
The equipartition theorem is an idea we're borrowing from statistical thermodynamics to predict expressions for the internal energy U for ideal gases. Many textbooks embed this concept with gases but Engel-Reid has squirreled it away in Chapter 14. To restate, for each quadratic term in the classical energy expression for a given molecule, there is a contribution of 1/2 kT to the average molecular kinetic energy. Implementing this rule is straightforward for any given structure if we just remember that each translational and rotational degree of freedom contributes 1/2 kT and each vibrational mode contributes a full kT.
As mentioned in class, the equipartition theorem ultimately fails when applied to gases that are nonideal -- of course, applying it to something like liquids or solids is heresy. Moreover it is inadequate when quantum effects are important, for example when the temperature is too low to significantly activate vibrational modes or if particular bonds are too stiff to vibrate at normal temperatures.
For ideal gases, we found that U=U(T); that is, internal energy is function only of temperature for a given sample. In fact it is a linear function of T, meaning that U = (constant)T. Since the heat capacity at constant volume, CV, is the derivative with respect to T, it will always be a constant for an ideal gas. The same can be said for CP, which should be apparent from the relationship CP = CV + nR (or, in molar form, CP,m = CV,m + R). I pulled this relationship out of thin air because we need it now but it will be proven when we are knee-deep in Chapter 3. Again, it is true only for ideal gases and applying it to any other system would be incorrect. Do not fret because more complicated systems (in fact, every system) will be addressed after we are finished laying down the mathematical foundations in Chapters 2 & 3.
A few notes on Problem 02 on hw.2 [P2.6 in Engel-Reid]: The notation for CP,m probably looks weird -- unfortunately it is becoming the standard way to write such equations in textbooks. Here's how I might write the same function:
Lastly, are you now able to predict CP,m for an ideal diatomic gas absorbed onto the surface of a metal (so that it is constrained to two dimensions)?
As mentioned in class, the equipartition theorem ultimately fails when applied to gases that are nonideal -- of course, applying it to something like liquids or solids is heresy. Moreover it is inadequate when quantum effects are important, for example when the temperature is too low to significantly activate vibrational modes or if particular bonds are too stiff to vibrate at normal temperatures.
For ideal gases, we found that U=U(T); that is, internal energy is function only of temperature for a given sample. In fact it is a linear function of T, meaning that U = (constant)T. Since the heat capacity at constant volume, CV, is the derivative with respect to T, it will always be a constant for an ideal gas. The same can be said for CP, which should be apparent from the relationship CP = CV + nR (or, in molar form, CP,m = CV,m + R). I pulled this relationship out of thin air because we need it now but it will be proven when we are knee-deep in Chapter 3. Again, it is true only for ideal gases and applying it to any other system would be incorrect. Do not fret because more complicated systems (in fact, every system) will be addressed after we are finished laying down the mathematical foundations in Chapters 2 & 3.
A few notes on Problem 02 on hw.2 [P2.6 in Engel-Reid]: The notation for CP,m probably looks weird -- unfortunately it is becoming the standard way to write such equations in textbooks. Here's how I might write the same function:
CP,m = 20.9 + 0.042T ( T in K, CP,m in J K-1 mol-1)It is important for you to understand why this gas is necessarily nonideal, despite the fact that the question states otherwise. In fact, for reasons stated above, it is not even possible to calculate a value for ∆U, which is also important for you to understand.
Lastly, are you now able to predict CP,m for an ideal diatomic gas absorbed onto the surface of a metal (so that it is constrained to two dimensions)?
Quiz 2
I'm looking at the schedule and am thinking about moving quiz 2 from October 12 [Friday] to October 15 [Monday]. Would that be met with approval, annoyance or the same grey ambivalence you have towards this blog?
Monday, October 1, 2007
Enthalpy
In the half hour before today's heartwarming quiz, we explored two conditions that change the First Law into simple statements about heat transfer. First, under constant volume conditions, in which no work other than PV-work is possible, the infinitesimal change in the internal energy dU was shown to equal dqV.
If we define a new thermodynamic function, called the enthalpy, as H=U+PV, we quickly find, under constant pressure and reversible conditions, that its infinitesima l change is equal to dqP. These relations allow us to further connect the heat capacities CP and CV to H and U respectively. This is only the first of many instances in which we will see the pressure|enthalpy and volume|internal energy links this quarter.
l change is equal to dqP. These relations allow us to further connect the heat capacities CP and CV to H and U respectively. This is only the first of many instances in which we will see the pressure|enthalpy and volume|internal energy links this quarter.
A comment on where we are so far. I am attempting to demonstrate how thermodynamics can be systematically used to calculate work and heat transfer, for any process, before moving onto their directionality. Since work and heat transfer are not directly measurable, we need some way to infer what's going on using directly measurable properties, like pressure, volume and temperature. We'll find out that these variables are not sufficiently rich to fully describe thermodynamic behavior, so we'll soon add other functions, like entropy and gibbs energy, to add to our growing toolbox.
If we define a new thermodynamic function, called the enthalpy, as H=U+PV, we quickly find, under constant pressure and reversible conditions, that its infinitesima
 l change is equal to dqP. These relations allow us to further connect the heat capacities CP and CV to H and U respectively. This is only the first of many instances in which we will see the pressure|enthalpy and volume|internal energy links this quarter.
l change is equal to dqP. These relations allow us to further connect the heat capacities CP and CV to H and U respectively. This is only the first of many instances in which we will see the pressure|enthalpy and volume|internal energy links this quarter.A comment on where we are so far. I am attempting to demonstrate how thermodynamics can be systematically used to calculate work and heat transfer, for any process, before moving onto their directionality. Since work and heat transfer are not directly measurable, we need some way to infer what's going on using directly measurable properties, like pressure, volume and temperature. We'll find out that these variables are not sufficiently rich to fully describe thermodynamic behavior, so we'll soon add other functions, like entropy and gibbs energy, to add to our growing toolbox.
Subscribe to:
Comments (Atom)